|
HOT SPRINGS
Analyses of the Waters of The Hot Springs of Arkansas Geological Sketch of Hot Springs, Arkansas |

|
THE CHEMICAL COMPOSITION OF THE WATERS OF THE HOT SPRINGS OF ARKANSAS.
METHODS OF EXAMINATION
Temperature.—The temperature of each spring was taken with an accurately standardized maximum thermometer on the date of the sanitary analysis of the water. Finally the temperatures of all of the springs were taken in one day. It will be noticed that these temperatures sometimes vary quite a few degrees for the same spring. This seems to be due to two causes. In the first place the temperature of the spring as it issues from the earth varies slightly from time to time; secondly, the springs sometimes have quite large basins, so that we can not get the temperature just as the water issues from the earth, but must take it as influenced by a comparatively large body of water, which in turn has been cooled to some extent by standing in the air. When these springs have recently been drained the temperature is nearly the same as where they issue from the earth, but when the basin is full the temperature is quite a few degrees lower.
Flow.—The flow of each spring was measured by observing the length of time taken to fill a vessel of known capacity from a pipe that drained the spring in question. In some cases such determinations could not be made, so the flow of the springs was estimated by comparing them with other springs of known flow. Such estimations were made by the head waterman of the reservation, Mr. Ed Hardin, who by long experience had arrived at such a point that he could come very near the correct figure.
Hydrogen sulphide.—The test for the presence of hydrogen sulphide was made both by boiling a sample of water and noticing the smell, and by passing the vapors over a piece of lead acetate paper. In a few cases, as a check, an actual determination of the hydrogen sulphide by the method given in Sutton's Volumetric Analysis was made. This is as follows:
About 0.5 c. c. of n/10 iodine was measured into a 500 c. c. flask and the water under examination run in till the color of the iodine disappeared. Five c. c. of starch water was added and n/10 iodine run in till the blue color appeared. The flask was then filled to the mark with distilled water. The amount of water actually titrated was found by subtracting the sum of iodine, starch solution, and distilled water from 500 c. c. As an excess of iodine solution was required to produce the blue color, a correction was applied by making 5 c. c. of starch solution up to 500 c. c. with distilled water and adding n/10 iodine until the color of the solution was just as blue as that in the actual determination. This figure subtracted from the first figure would give the number of c. c. of n/10 iodine used by the hydrogen sulphide. In every case tried the correction was just equal to the original figure, and in neither of the other tests was hydrogen sulphide found to be present in any of the springs.
Nitrogen and oxygen.—Nitrogen and oxygen were determined by making use of the Tiemann and Preusse modification of Reichhardt's apparatus, the description of which is here taken from Hempel's Gas Analysis (translated by L. M. Dennis, Cornell University):
This consists of two flasks, A and B (Fig. 1), each of about 1 liter capacity and connected by tubes with the gas collector C. The flask A is fitted with a perforated rubber stopper in which is inserted the glass tube a bent at a right angle and ending flush with the lower surface of the stopper; a is joined by a piece of rubber tubing to the tube bc, which in turn connects with the gas collector C. C is held by a clamp, has a diameter of 30 mm., is about 560 mm. long, and at the upper end is drawn out to a short, narrow tube, which can be closed with the rubber tube and pinchcock g. In the lower end of C is a rubber stopper with two holes through one of which the tube bc, projecting about 280 mm. into C, is inserted. Through the other opening passes the tube d, which extends only slightly beyond the stopper and connects C with the flask B. B has a double bore rubber stopper carrying the tubes e and f; e ends about 10 mm. above the bottom of the flask and above the stopper it is bent at a right angle and is connected with d. The tube f, which need not project below the stopper, carries a thin rubber tube X about 1 meter in length and is provided with a mouthpiece. A pinchcock for closing the rubber between a and b is also needed.
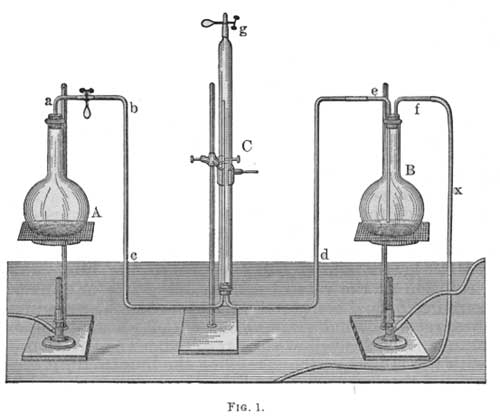
|
| FIG. 1. |
The apparatus thus arranged is made ready for a determination by filling the flask B somewhat more than half full of boiled, distilled water and removing the flask A by slipping the tube a out of the rubber connection; then by blowing into the rubber tube X, water is driven over from the flask B into the gas collector C and the adjoining tubes until the air is wholly displaced. The rubber tubes at b and g are now closed with pinchcocks. The flask A is then filled to the brim with distilled water, the stopper is inserted, water being thereby driven into the tube a and the flask is again connected with b, the pinchcock being opened.
The water in B is now heated to gentle boiling, and that in A is allowed to boil somewhat more rapidly. The absorbed air is thus driven out, and the gases dissolved in the water which is in A and C collect in the upper part of C, from which they are removed by occasionally opening the pinchcock at g and blowing into the rubber tube X.
When upon cooling the apparatus, the
gases which have collected disappear, the heating of the flask A is
discontinued, the pinchcock between a and b is closed and
A is disconnected and emptied. The water in C and B is now entirely free
from absorbed gases and air can not enter from without, because the
liquid in B is kept continually boiling. The apparatus is now ready for
a determination, which is made as follows: The cooled flask A, whose
capacity has been previously determined, is filled with the water to be
examined and the stopper is pressed in so far that the air in the tube
a is completely driven out; a is then
connected with b, care being taken that in so doing no air
bubbles are inclosed. The pinchcock between a and b is
opened and the water in A is heated to gentle boiling. The dissolved
gases are hereby driven over into the gas collector C. Steam is formed
at the same time. The heating of the flask A must be so regulated that
the gas and steam evolved never drive out more than half the liquid in
C, otherwise there is danger of gas bubbles entering the tubes d
and e and thus escaping.
After heating for about 20 minutes the flame under A is removed. In a few minutes the steam in A and C condenses, and water passes from B to C and A. If a gas bubble is observed in A which will not disappear when the neck of A is cooled by applying a wet towel two or three times, the flask A must again be heated and cooled in the manner just described. The operation is ended when the hot liquid flows back and completely fills A.1 The rubber tube g is then connected with a small piece of thermometer tube which is filled with water, and gas standing over the hot liquid in C is driven over into a modified Winkler gas burette by blowing into the tube X and opening the pinchcock g.
1It has been observed in waters rich in bicarbonates that it is nearly impossible to drive off all the CO2 by this means, but the O and N and part of the CO2 are driven off in the course of a half hour's boiling. Therefore the author did not continue boiling A, even though a small bubble of gas was present, more than one-half an hour.
The gases in the burette were allowed to cool for about 10 minutes, and then passed into a simple absorption pipette filled with potassium-hydrate solution (one part KOH to two parts of water). The pipette was shaken two or three times to absorb the carbon dioxide, and the residual gases passed back into the burette. The burette was allowed to stand for a few minutes and the volume of the gas read off. This gave the volume of oxygen + the volume of nitrogen. The gas was then passed into a double-absorption pipette filled with potassium pyrogallate, prepared by mixing 5 grams pyrogallic acid and 15 c. c. of water with 120 grams of potassium hydroxide and 80 c. c. of water. After being shaken with this solution for about four minutes the gas was passed back into the burette, the burette allowed to stand for a few minutes, and the reading taken. The last reading gave the number of c. c. of nitrogen present, and the difference between the first and last reading; the number of c. c. of oxygen. A temperature and barometric pressure reading were also taken, to correct the gas volume to 0° C. and 760 mm. pressure. Numerous precautions as to temperature, saturation of reagents, etc., not mentioned in the above brief sketch, were taken, all of which can be found in any standard work on gas analysis.
Carbon dioxide (in excess of that necessary to form normal carbonates).—The determination of the carbon dioxide existing in water in excess of that present as normal carbonates was made by a method given in Sutton's Volumetric Analysis and designed by Pettenkofer. One hundred c. c. of the water was treated in a flask with 3 c. c. of a saturated solution of calcium chloride, 2 c. c. of a saturated solution of ammonium chloride, and 45 c. c. of a saturated solution of calcium hydroxide, whose strength had previously been determined in terms of n/10 hydrochloric acid, using lacmoid as indicator. The flask was stoppered, the solution well mixed, and the whole set aside for 12 hours to allow the calcium carbonate to settle. At the end of this time 50 c. c. of the clear solution was drawn off in a pipette and titrated with n/10 hydrochloric acid, using lacmoid as indicator. This result was multiplied by three and subtracted from the amount of n/10 hydrochloric acid necessary to neutralize 45 c. c. of the calcium hydroxide solution, thus giving the amount of calcium hydroxide solution that had been acted on by the carbon dioxide in terms of n/10 acid. Multiplying the number of c. c. so found by 0.0022, the weight of carbon dioxide in 100 c. c. above that necessary to form normal carbonates was found. Dividing the weight so found by the weight of 1 c. c. of carbon dioxide at 0° C. and 760 mm. pressure and multiplying the result by 10, the number of c. c. of carbon dioxide in a liter in excess of that necessary to form normal carbonates was given.
Carbon dioxide (given off from the bicarbonates when they are evaporated to dryness).—In making this determination the method of Cameron1 for the "Estimation of carbonates and bicarbonates in aqueous solution" was used. By this method the amount of bicarbonic acid ion (HCO3) was determined, and from this we could easily estimate how much of the bicarbonic acid would remain as the normal carbonate and how much be given off as carbon dioxide. The method is as follows:
To 100 c. c. of the water was first added a few drops of phenolphthalein. In case there were alkali carbonates present the usual red color would be evident. The solution was now titrated with a solution of HKSO4, containing 6.758 grams to the liter, adding the HKSO4 solution at the rate of a drop every two or three seconds, until the red color had completely disappeared. The reading on the burette was recorded, and to the clear solution was added one drop of methylorange. A pure yellow color resulted. The titration was continued with the HKSO4 without refilling the burette until the change to a very slightly darker and reddish color was noted. The change was faint and required practice to detect. The reading at this point was also recorded.
The first reading recorded gives the amount of alkali carbonates present and must be multiplied by the factor 0.002979 for the result in grams of CO3 ions.
For the number of grams of HCO3 ions present the first recorded reading is multiplied by two and the result subtracted from the second reading, and this remainder is multiplied by the factor 0.003028. In no case were carbonates found in any of the springs by the above method, but only bicarbonates.
Having now obtained the weight of HCO3 ions in 1,000 c. c. of water, we next calculate the weight of CO2 given off when a like volume is evaporated to dryness, and dividing this result by the weight of 1 c. c. of carbon dioxide at 0° C. and 760 mm. pressure the number of c. c. of carbon dioxide given off from the bicarbonates is the result. Subtracting the number of c. c. of carbon dioxide given off from the bicarbonates from the number of c. c. of carbon dioxide in excess of that necessary to form normal carbonates, we have left the number of c. c. existing in solution in a free state.
1Report 64 U. S. Department of Agriculture; American Chemical Journal. 22,471 (1900).
Bicarbonic acid.—The amount of bicarbonic acid present in the spring was estimated during the process of determining the amount of carbon dioxide given off from bicarbonates in the paragraph above.
It will be noticed in several of the analyses of the different springs that the amount of carbon dioxide (set free from bicarbonates on evaporating to dryness) and calculated from the bicarbonic acid does not agree with the amount of bicarbonic acid found in solution. This is because the samples for determining the carbon dioxide and bicarbonic acid were taken at widely different periods, and the amount of bicarbonic acid had evidently changed somewhat during the intervening time. This is easily explained when we remember that many of the springs are supplied from two or three different spring heads, which doubtless vary from time to time both in their amount of flow and in the amount of bicarbonic acid held in solution.
Nitric acid.—For the determination of nitric and nitrous acid, free and albuminoid ammonia, and oxygen consuming capacity the methods as given in Mason's Examination of Water were followed in all their principal details. In determining nitric acid, 100 c. c. of the spring water was treated with 2 drops of a saturated solution of sodium carbonate and evaporated to dryness on the water bath. The residue was treated with 2 c. c. of phenol sulphonic acid (made by mixing 148 c. c. of pure sulphuric acid, 12 c. c. of water, and 24 grams of phenol), a little water added, and then an excess of ammonia. The solution was transferred to a 100 c. c. Nessler jar, the volume made up to 100 c. c. with distilled water, and the depth of the yellow color compared with that produced by treating different measured amounts of standard potassium nitrate (containing 0.01 milligram of nitrogen as nitrate in each c. c.) in the same manner.
Nitrous acid.—For this determination 100 c. c. of the water was placed in a 100 c. c. Nessler jar and treated with 1 drop of concentrated hydrochloric acid. One c. c. of sulphanilic acid (containing 1 gram in each 100 c. c. of water) was then added, followed by 1 c. c. of a solution of napthylamine hydrochloride (obtained by boiling 0.5 gram of the salt with 100 c. c. of water for 10 minutes at constant volume), and the whole well mixed. The Nessler jar was then set aside for half an hour, along with several other Nessler jars containing known amounts of a standard nitrite solution (containing 0.0001 milligram of nitrogen as nitrite in each c. c.), made up to 100 c. c. with nitrite-free water, and treated with hydrochloric acid, sulphanilic acid, and napthylamine hydrochloride in the manner just described. By comparing the depth of pink color in the known and unknown solutions the amount of nitrite could be determined.
Free ammonia.—A large flask of about 1-1/2-liter capacity was connected to an upright bulbed condenser by means of a rather large glass tube and soft, new, rubber-stopper connections. In this was placed 5 c. c. of a saturated solution of sodium carbonate and 200 c. c. of ammonia-free water. The water was distilled off in 50 c. c. Nessler jars until no more ammonia was shown, when the jars were nesslerized. Five hundred c. c. of the water under examination was now added and the distillation in 50 c. c. Nessler jars continued till ammonia ceased to be given off. About four or five jars were usually necessary. These jars were nesslerized and the depth of color compared with that in other jars which contained known amounts of a standard ammonium chloride solution (containing 0.01 milligram of NH3 in each c. c.), made up to 50 c. c. with ammonia-free water and nesslerized in the same manner.
Total ammonia.—The same apparatus was used as that mentioned in the paragraph above. In it were placed 200 c. c. of distilled water and 50 c. c. of alkaline permanganate solution (prepared by dissolving 200 grams of potassium hydroxide and 8 grams of potassium permanganate in 1,250 c. c. of water and boiling the whole down to about l liter). The water was distilled off in 50 c. c. Nessler jars till ammonia ceased to come over. Five hundred c. c. of water under examination was now added and the distillation continued till ammonia ceased to come off. Six jars were in all cases sufficient. These jars were nesslerized and compared with nesslerized jars of known strength just as in the determination of free ammonia. From the total ammonia thus found subtract the free ammonia and the result is the albuminoid ammonia in 500 c. c. of water.
Many precautionary details of the two above methods are not given, but can be found by consulting any good book on water analysis.
Oxygen-consuming capacity.—In making this determination two solutions were first prepared: (1) A standard solution of potassium permanganate containing 0.3952 gram to the liter, each c. c. of which has 0.1 milligram of oxygen available for oxydation; and (2) a standard solution of oxalic acid containing 0.7875 gram of crystallized oxalic acid to the liter. The value of the oxalic acid in terms of the permanganate was determined by boiling 10 c. c. of oxalic-acid solution and 200 c. c. of distilled water with 10 c. c. of sulphuric acid (1—3) and titrating the fluid while boiling with the standard permanganate solution to the appearance of a pink color. In the actual determination 200 c. c. of the water in a porcelain dish was treated with 10 c.c. of sulphuric acid (1—3) and the whole brought to the boiling point. Standard permanganate was run in until the water was quite red and the boiling continued for 10 minutes, adding permanganate every now and then to keep the pink color about the same. The boiling was now stopped, 10 c. c. of oxalic acid run in, which destroyed the color, and the solution titrated with the standard permanganate to the appearance of a pink color. From the total number of c. c. of permanganate used was subtracted the number of c. c. equal to 10 c. c. of oxalic acid. The result gives the number of c. c. of permanganate required for 200 c. c. of water.
Total solids.—Measured amounts of the water were evaporated to dryness in weighed platinum dishes on the steam bath. The dishes were dried for 12 hours at the temperature of boiling water, cooled in the desiccator, and weighed. The increase in weight of the dish gives the amount of solids present in the volume of water used.
To determine chlorine, iron, and aluminum, manganese, bromine, iodine, arsenic, and boric acid large quantities of the water were evaporated to dryness after the addition of a small amount of sodium carbonate. The residue thus obtained was boiled with distilled water, transferred to a filter, and thoroughly washed with hot water. The residue in the paper was dried and transferred to the dish in which the evaporation was made, the paper burned and added, and the whole kept for the determination of iron, aluminum, and manganese. The filtrate was made to a definite volume and aliquot portions taken to determine the constituents mentioned above other than iron, aluminum, and manganese.
Chlorine.—An aliquot portion from the above filtrate was treated with a few drops of phenolphthalein and n/10 HKSO4 added at the rate of a drop every few seconds until the red color had entirely disappeared, thus showing that all of the carbonates had changed to bicarbonates.1 A few drops of potassium chromate indicator were then added and the chlorides titrated with a solution of silver nitrate each c. c. of which would precipitate l milligram of chlorine.
lSee Cameron's paper in Amer. Chem. Journal, 23, 481, 1900.
Iodine and bromine.—The qualitative tests for the presence of iodine and bromine were very much the same as those used in Fresenius. Another aliquot portion from the above filtrate was evaporated to dryness on the steam bath. Two or 3 c. c. of water were added to dissolve and soften up the residue and enough absolute alcohol added to bring the percentage of alcohol down to about 90 per cent. This was boiled and filtered and the treatment with 90 per cent alcohol repeated once or twice. Two or 3 drops of sodium hydrate solution were added to the filtrate and it was evaporated to dryness. The same process of extracting with 90 per cent alcohol was repeated on the new residue and the extract filtered off from the undissolved portion. A drop of sodium hydrate was added to the filtrate and it was evaporated to dryness. The residue was treated with a little distilled water, dilute sulphuric acid added to acid reaction, the liquid transferred to a test tube, and a little carbon disulphide added. Three or 4 drops of potassium nitrite solution were then added and the test tube shaken. The presence of iodine was shown by a pink color in the carbon bisulphide. Chlorine water was then added until the pink color due to the iodine had disappeared, then a little more chlorine water.
The presence of bromine was shown by an orange color in the carbon bisulphide.
In no case did a sample of spring water give nearly as distinct a reaction for iodine and bromine as did a known sample of water containing 0.2 milligram of both iodine and bromine, as iodides and bromides, to the liter.
An attempt was made to determine iodine and bromine quantitatively in spring No. 15 by evaporating down a large volume of water, but the attempt failed because both these elements were present in such minute traces. The method used was the same as that described by Gooch and Whitfleld2 and is as follows: The iodides and bromides were extracted with 90 per cent alcohol in the same manner as described above.
2Bulletin 47 of U. S. Geological Survey.
The alcohol extract was evaporated to dryness, acidulated with dilute sulphuric acid, mixed with a ferric sulphate solution, and distilled from a retort which was joined to a condenser sealed by a U-tube filled with water and carbon bisulphide. If a very small amount of iodine had been present it would have colored the carbon bisulphide and could have been titrated with sodium thiosulphate, but not enough was present.
After the distillation had been continued long enough to be sure that all iodine had been volatilized, cyrstals of potassium permanganate were added and the distillation continued the same as before, except that the U-tube acting as a seal was now filled with water and chloroform. The contents of the tube were treated with sodium hydroxide and zinc in a breaker and the chloride and bromide solution so formed acidified with nitric acid and precipitated with silver nitrate. The precipitate was dried and weighed. It was then dissolved in potassium cyanide and the silver precipitated by electrolysis.1 In this way data on the weight of the combined silver chloride and bromide and the weight of the silver in same was determined. From this the weight of the bromine could be calculated, which in this case was nothing.
lAmerican Chemical Journal, vol. 8, p. 421.
Arsenic acid.—An aliquot portion of the above filtrate was acidified with hydrochloric acid, the solution heated to 700 C., and a current of hydrogen sulphide passed through for several hours. In case either arsenic, copper, or lead were present they would be precipitated. No precipltation took place in any of the springs.
Boric acid.—A test for boric acid was made in the following manner: A part of the above filtrate was evaporated to dryness, treated with a cubic centimeter or two of water, and slightly acidified with hydrochloric acid. About 25 or 30 c. c. of absolute alcohol was added, the solution boiled, and filtered. This was repeated. The filtrate was made slightly alkaline with sodium hydrate and evavorated to dryness. A very little water was added, the solution sightly acidified with hydrochloric acid, and a strip of turmeric paper placed in the liquid. The whole was evaporated to dryness on the steam bath, and the heating continued until the turmeric paper had become entirely dry. In case boric acid were present the turmeric paper took on a cherry-red color.
A quantitative determination of boric acid was made in the case of two springs to serve as an example of all the other springs.
The method used was the same as that described by Gooch,2 except that a slightly different form of apparatus was used.
2American Chemical Journal, vol. 9, p. 23.
The apparatus used by the author (Fig. 2) consisted of a round-bottomed flask with a constricted neck joined to an upright bulbed condenser by means of a glass tube slightly sloping toward the flask instead of being bent at right angles. The flask was heated by being immersed in a paraffin bath, and the distillate was received in a small flask joined to the condenser by means of a grooved cork. The method was as follows:
An aliquot portion of the above filtrate evaporated to dryness was slightly acidified with acetic acid and transferred to the round-bottomed flask, 10 c. c. of methyl alcohol was added, the flask lowered in the paraffin bath, and distilled to dryness at a temperature of 130° C. to 140° C., collecting the distillate in the flask attached to the condenser with a grooved stopper. The paraffin bath was lowered, the flask allowed to cool, and 10 c. c. more of mythyl alcohol added. This was then distilled over and the same process repeated six times, except that after the fourth time a couple of drops of acetic acid were added. A large platinum crucible now received about l gram of quicklime and was blasted until it ceased to lose weight. The constant weight was recorded and the distillate transferred to the crucible. The alcoholic solution of boric acid and the quicklime were stirred together for about 15 minutes with a platinum rod to be sure that all boric acid was fixed.
The volatile contents of the crucible were now evaporated off at a low temperature. It was found necessary to grease the edges of the crucible with vaseline to keep the solution from crawling over. After the contents of the crucible had been evaporated to dryness the crucible was fully dried in the air bath and finally blasted. The increase in weight of the crucible gives the weight of boric anhydride (B2O3) present.
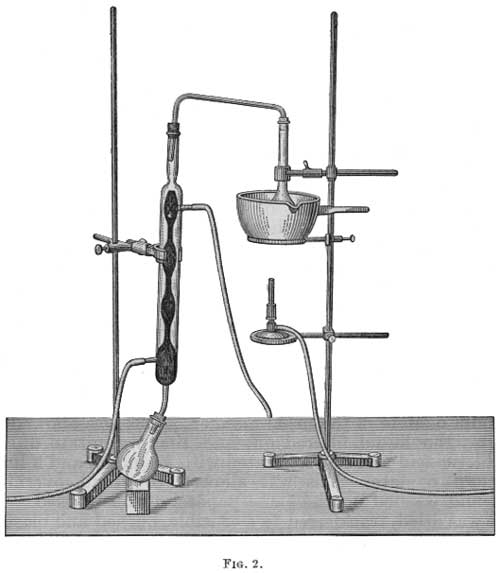
|
| FIG. 2. |
Iron, aluminum, and manganese.—The residue spoken of previously that was reserved for the determination of iron, aluminum, and manganese was treated with hydrochloric acid and evaporated to dryness. It was thoroughly dried at about 120° C., again taken up with water and hydrochloric acid, and filtered. The filtrate was evaporated to dryness and dried at 120° C. It was then taken up with hydrochloric acid and water and filtered again. This filtrate was heated to the boiling temperature, and ammonia added, a drop at a time, until it could be very faintly smelled coming off from the solution. The solution was then filtered and the precipitate well washed with hot water, burned, and weighed as Fe2O3 and A12O3 in the ordinary manner.
The ammoniacal filtrate from above was treated with a few drops of bromine, more ammonia was then added, and the whole boiled after stirring up. The vessel was removed from the source of heat, cooled a little, and a little more bromine and ammonia added. This process repeated once or twice precipitated all the manganese as the oxide. The solution was made slightly acid with acetic acid, filtered, and washed at once with hot water. The filter and contents were burned and weighed as Mn3O4. This is the method by which the iron, aluminum, and manganese were determined in springs 24 to 46, inclusive. In the first 23 springs these three elements were determined in the same portion that was used for the estimation of calcium and magnesium.
Silica.—In this determination a large quantity of water was evaporated to dryness in platinum with the occasional addition of small amounts of hydrochloric acid. After all the water had been evaporated to dryness, the dish and contents were completely dried at 120° C. The residue was taken up with hydrochloric acid and water, heated and filtered, washing the residue thoroughly with hot water. This process took out most of the silica. The filtrate was then evaporated to dryness, dried thoroughly at 120° C., again taken up in hydrochloric acid solution by heat, and filtered. The filtrate was made to a definite volume, aliquot portions of which were used for the determination of calcium, magnesium, sulphuric acid, potassium, sodium, lithium, and phosphoric acid. The two residues were transferred to a crucible, burned and blasted in the ordinary way, and finally weighed as silica.
Calcium and magnesium.—An aliquot portion of the above filtrate was first treated with ammonia and filtered, then treated with ammonia and bromine water and filtered, and finally treated with ammonium oxalate in the usual manner. This was allowed to stand overnight, the liquid filtered off, and the precipitate dissolved in hydrochloric acid and reprecipitated with ammonia and a little extra ammonium oxalate. This was allowed to stand overnight and filtered and washed on the same paper previously used. The precipitate was dried, transferred to a crucible, burned and blasted in the ordinary way, and finally weighed as calcium oxide. The combined filtrates were evaporated to dryness in platinum and the major part of the ammonium salts driven off by the aid of heat. The residue was dissolved in dilute hydrochloric acid and filtered. The filtrate was made slightly ammoniacal, enough sodium phosphate solution added, a drop at a time, to precipitate all magnesium, and 10 c. c. of concentrated ammonia finally added, drop by drop. The beaker was covered and allowed to stand overnight, filtered, washed with dilute ammonia water, dried, blasted, and weighed as magnesium pyrophophate.
Sulphuric acid, potassium, sodium, and lithium.—Another portion of the above filtrate was precipitated while boiling with hot, dilute barium chloride and, after standing, filtered from the precipitated barium sulphate, which was washed, dried, burned; and finally weighed in the ordinary way.
The filtrate was evaporated to dryness and taken up with water. This solution was precipitated with a solution of barium hydrate and filtered off from the insoluble magnesium hydrate. The magnesium hydrate precipitate was well washed and the combined filtrate and washings treated with ammonia, ammonium carbonate, and a little ammonium oxalate to precipitate barium and calcium. This precipitate was allowed to stand overnight, filtered off, and well washed. The filtrate and washings were evaporated to dryness on the steam bath, dried, and all of the ammonium salts driven off by gentle heat. The residue was taken up with water, filtered through a small filter, using as little wash water as possible, evaporated to a small volume, and finally again precipitated with a drop of ammoma and two to three drops of ammonium carbonate and oxalate. If any precipitate appeared, which was not usually the case, it was filtered off and the same process repeated. In any case, the solution was filtered from the magnesium hydrate that had precipitated out on concentrating the solution. The filtrate was then evaporated to dryness and all ammonium salts driven off by heating in platinum to a little below redness. The residue was taken up with a little water and filtered through a small filter, again using as little wash water as possible, and again heated in platinum to a point slightly below red heat. By this time all of the magnesia should have been removed. The residue was then taken up with a little water, filtered into a weighed platinum dish, treated with a few drops of hydrochloric acid, and evaporated to dryness. This residue was thoroughly dried, heated to a little below redness, cooled in a desiccator, and finally weighed as the combined chlorides of potassium, sodium, and lithium.
The determination of lithium was then made according to the method of Gooch1—i. e., the combined chlorides were dissolved in water and transferred to a small beaker, where they were again evaporated nearly to dryness. About 30 c. c. of amyl alcohol was added and the contents of the beaker boiled until the temperature had risen to approximately the boiling point of the amyl alcohol, showing that all of the water had been driven off. The liquld was cooled slightly, and a drop of hydrochloric acid was added to reconvert small amounts of lithium hydrate to lithium chloride. The boiling was then continued to again drive off all water, until finally the liquid had reached a volume of about 15 c. c. The amyl alcohol was then filtered off in a weighed platinum dish and the filter washed with a little amyl alcohol that was also allowed to run into the dish. The amyl alcohol was driven off from the filter and beaker in the air bath and these two kept for the determinations of potassium and sodium. The contents of the platinum dish were evaporated to dryness, treated with a little dilute sulphuric acid, and finally burned and weighed. This gave the weight of the lithium sulphate, from which was subtracted 0.0017 gram to correct for the solubility of the sodium and potassium chlorides in the amyl alcohol. The residue was finally tested with the spectroscope for the lithium line. In every case the lithium line was found, but in no case was any lithium sulphate left after applying the correction of 0.0017 gram. The lithium was therefore reported as traces.
lAmerican Chemical Journal, vol. 9, p. 33.
The contents of the beaker and filter from which the amyl alcohol had been driven were then used for the determination of potassium and sodium. The contents of the beaker were dissolved in hot water and passed through the filter, which was thoroughly washed. The combined filtrate and washings were transferred to a porcelain dish, treated with platinum chloride solution, and evaporated nearly to dryness. The residue was treated with 80 per cent alcohol and thoroughly washed on the filter with this medium until all platinum chloride had been washed out. The filter paper was dried at the temperature of boiling water, and the residue dissolved in water and passed into a weighed platinum dish from which the water was evaporated off, the dish and contents dried at the temperature of boiling water, and finally weighed as potassium platinic chloride. An addition of 0.0008 gram of potassium chloride to the weight of this substance found is necessary.
The weight of the sodium chloride is found by subtracting the combined weights of the lithium chloride (in this case nothing) and the potassium chloride (corrected) from the total weight of the three chlorides.
Of course if the amyl alcohol in the determination of lithium above is not evaporated to exactly 15 c. c. the corrections will be different from those mentioned above.1
lFor the discussion of this, see the original article already mentioned.
. Phosphoric acid.—A third aliquot portion from the filtrate mentioned above was treated with about 10 c. c. (con.) nitric acid and evaporated in a porcelain dish nearly to dryness to drive off hydrochloric acid. The residue was taken up with water and if necessary filtered. Ammonia was added to alkalinity and then nitric acid to just bring back to acidity. Some ammonium nitrate was added and the beaker heated in the water bath to 45° to 50° C. Molybdate solution was then added and the solution kept at a temperature of 45° to 50° C. for half an hour. The yellow precipitate formed at this point appeared in most cases only in traces, but in a few cases it was filtered off and washed with cold water till it was entirely free of nitric and molybdic acids. The precipitate and filter were then transferred to a beaker, a little water added, and the paper and contents thoroughly beaten into a pulp. The yellow, precipitate was then dissolved by the addition of a small amount of standard potassium hydroxide solution (1 c. c. = 1 milligram of P2O3); phenolphthalein was added and the solution titrated with standard nitric acid solution of exactly the same strength as the alkaline solution. From the data so obtained the amount of phosphoric acid ion in the water can be calculated.2 For the determination of fluorine the same method was used as described by Gooch and Whitfield.3 For the determination of barium and strontium a combination of Gooch and Whitfield's method along with another was employed. They are briefly as follows:
2Bul. 46 (revised edition), U. S. Department of Agriculture, Division of Chemistry. 1899.
3Bul. 47, U. S. Geological Survey.
Fluorine.—A large quantity of water was evaporated to dryness and filtered off from the residue which was washed on the filter. The filter and contents were dried, the contents placed aside and the filter burned and the ash added to the contents. The whole was now transferred to a flask, which was so arranged as to allow a current of air to pass through any liquid that might be in the bottom, and from there into an attached U-tube, partly filled with dilute ammonia. Concentrated sulphuric acid was added to the contents of the flask, and a current of dry air passed through the liquid, and from there into the U-tube. The flask was heated to 150° C. If any considerable amount of fluorine had been present it should have been volatilized as silicon tetrafluorid and then decomposed by the dilute ammonia in the U-tube, depositing silica in so doing. No silica appeared at this point in the spring examined. The contents of the U-tube was removed and treated with zinc oxide dissolved in ammonia, evaporated till ammonia ceased to come off, and filtered. The filtrate was treated with calcium chloride, followed by sodium carbonate in boiling solution, filtered, and washed. The residue was ignited and extracted with acetic acid. Operating in this way no residue of calcium fluoride was found.
Barium and strontium.—The residue left in the flask from the above determination was transferred to platinum, treated with enough hydrofluoric acid to volatilize all silica and with some sulphuric acid and evaporated to dryness. This treatment was repeated. The residue was fused with sodium carbonate, treated with water and a few drops of alcohol, filtered, and washed. The contents of the filter was digested with hot dilute acetic acid to dissolve barium, strontium, magnesium, and calcium carbonates, and filtered. The filtrate was then nearly neutralized with ammonia and about 50 times the weight of the combined sulphates in ammonium sulphate was added, which ammonium sulphate was dissolved in 4 times its weight of water. The whole was allowed to stand overnight. In case barium or strontium were present they would be precipitated here as the sulphates. Only a slight nonweighable opalescence appeared, however, in the spring examined. For the sake of completeness, and to be able to test the final residue with the spectroscope, the process was carried on just as in an actual determination. The precipitated sulphates were filtered and washed with a concentrated solution of ammonium sulphate till no more calcium was present in the wash water, as shown by the ammonium oxalate test. The filter was ignited and the residue evaporated to dryness with a drop or two of sulphuric acid. The combined sulphates so obtained from a very large quantity of water did not weigh over 0.5 milligram, and most of this was calcium sulphate. The extremely small residue was fused with sodium carbonate, treated with a very small quantity of water, and filtered on a very small filter paper, washing only once. Dilute hydrochloric acid was now passed through the filter and the filtrate containing any barium and strontium as the chlorides was collected in a platinum dish and evaporated to dryness. The minute residue was tested by the spectroscope for the barium and strontium lines both of which were faintly seen.
Strontium.—This substance was determined in .a separate portion. The oxide of calcium, which had been obtained by blasting the ammonium oxalate precipitate in the determination of calcium, was transferred to a small flask and dissolved in concentrated nitric acid. The acid was entirely evaporated off by means of a current of air and heating in a paraffin bath to 135° C. The flask and contents were dried at 140° C., and the completely dried nitrates were treated with the least possible quantity of a mixture of equal parts of absolute alcohol and ether, necessary to dissolve the calcium nitrate. The flask was corked, allowed to stand over night, and the insoluble residue, if any, filtered off on the smallest possible filter and washed with the ether-alcohol mixture. The strontium nitrate on the filter was washed with water into a platinum dish and evaporated to dryness. The dish was blasted to change the nitrate to the oxide. No increase in the weight of the dish was noticed, yet upon treating the contents of the dish with a little hydrochloric acid, evaporating nearly to dryness and testing with the sectroscope, the strontium lines were seen.
| <<< Previous | <<< Contents>>> | Next >>> |
haywood-weed/sec2.htm
Last Updated: 22-Dec-2011